MedicalResearch.com Interview with:
[caption id="attachment_50605" align="alignleft" width="181"]

Upekha Liyanage[/caption]
Upekha Liyanage MBBS | PhD Student
School of Medicine | University of Queensland
Statistical Genetics Laboratory
QIMR Berghofer Medical Research Institute
MedicalResearch.com: What is the background for this study? What types of skin cancers are linked to these genes?
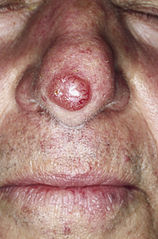
Response: Basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) collectively referred to as “keratinocyte cancers” are the commonest forms of cancers of the skin. Although these cancers are less aggressive than melanoma, due to their large numbers, they pose a significant burden to the healthcare expenditure. Also, these cancers are relatively understudied when compared with melanoma. Notably, BCC and SCC are not routinely reported in cancer registries. In Australia, Medicare data are used to estimate the incidence and costs associated with these cancers. Expenditure in Australia for the diagnosis, treatment and pathology, almost exceeds $700 million for both BCC and squamous cell carcinoma. In Unites States, the average annual cost for skin cancer including melanoma is approximately $8.1 billion.
Previous research has led to identification of 29 BCC and 11 squamous cell carcinoma genetic risk variants and 7 of them overlap with both BCC and SCC risk. So, to strengthen the preventive efforts and to reveal new therapeutics, it is very timely and critical to explore more on the genetic susceptibility of these deadly cancers. We analysed ~48,000 cancer cases with ~630,000 skin cancer free controls from European ancestry population in Australia, UK and USA.