
08 Jun Hebrew University: First WWOX Gene Replacement Therapy Administered to Child With Hereditary Seizures
[caption id="attachment_74143" align="aligncenter" width="500"] Conceptual illustration of AAV9-mediated delivery of the WWOX gene to neurons, representing the first clinical use of a gene replacement therapy designed to restore WWOX function in the brain of an infant with WOREE syndrome.Credit: Hebrew University of Jerusalem / AI-generated illustration[/caption]
MedicalResearch.com Interview with:
Prof. Rami Aqeilan
Jacob M. Eisenberg and Thomas W. Baylek Chair for Medical Research in the field of Genetic Engineering
Lautenberg Center for Immunology and Cancer Research
Faculty of Medicine
Hebrew University of Jerusalem
Jerusalem, Israel
This therapy is based on more than a decade of research led by Prof. Rami Aqeilan, brought together with scientists, clinicians, and biotechnology leaders from Israel and the United States, including Dr. Naama Orenstein and Dr. Dror Kraus of Schneider Children's Medical Center and Dr. Yael Weiss, CEO of Mahzi Therapeutics.
MedicalResearch.com: What is the background for this study? Would you briefly explain the functions of the WWOX gene? Response: WWOX (WW domain-containing oxidoreductase) is a highly conserved gene that plays essential roles in brain development, neuronal function, and cellular stress responses. Nearly two decades ago, our laboratory and others began studying WWOX because of its involvement in cancer biology. However, over the past decade it became increasingly clear that WWOX is also critical for normal brain development. Inherited loss-of-function mutations in WWOX cause a devastating neurological disorder known as WOREE syndrome (WWOX-Related Epileptic Encephalopathy). Affected children typically develop severe, treatment-resistant epilepsy during infancy, profound developmental delay, intellectual disability, and significant motor impairment. Unfortunately, there has been no disease-modifying therapy available for these patients. The foundation for this therapeutic approach came from years of fundamental research in our laboratory aimed at understanding the biological role of WWOX in the nervous system. Using genetically engineered mouse models, we discovered that deleting WWOX specifically in neurons was sufficient to reproduce the major neurological features observed in mice lacking WWOX throughout the entire body. This finding demonstrated that neuronal WWOX deficiency is a primary driver of the disease and suggested that restoring WWOX function in neurons might be sufficient to achieve therapeutic benefit. Based on this insight, we developed a gene replacement strategy designed to restore WWOX expression selectively in neurons using an adeno-associated viral (AAV) vector. In preclinical studies, delivery of this vector into the brains of WWOX-deficient mice resulted in remarkable rescue of the disease phenotype. Treated animals exhibited normal behavior, elimination of seizures, and substantial correction of the neurological abnormalities associated with WWOX deficiency. These findings provided the critical proof-of-concept that neuronal gene replacement could effectively reverse key features of the disease and laid the scientific foundation for translating this approach toward clinical application in patients with WOREE syndrome.


 Dr. Tauscher-Wisniewski,[/caption]
Sitra Tauscher-Wisniewski, MD
Vice President Clinical Development & Analytics
Novartis Gene Therapies
MedicalResearch.com: What is the background for this study? Would you briefly describe the condition of Spinal muscular atrophy (SMA)?
Response: At the 2023 Muscular Dystrophy Association Conference, we presented new data from two of our Long-Term Follow-Up (LTFU) studies, LT001 and LT002, which show the continued efficacy and durability of Zolgensma across a range of patient populations, with an overall benefit-risk profile that remains favorable. LT001 is a 15-year ongoing observational LTFU study following the Phase 1 START patients, who were the very first patients to receive our gene replacement therapy. LT-002 is a voluntary Phase 4 15-year ongoing follow-up safety and efficacy study of Zolgensma IV and investigational intrathecal (IT) OAV101 in patients previously treated in the Phase 3 IV studies (STR1VE-US, STR1VE-EU, STR1VE-AP, SPR1NT) and the Phase 1 IT study (STRONG).
Spinal muscular atrophy (SMA) is a rare, devastating genetic disease that leads to progressive muscle weakness, paralysis, and when left untreated in one of its most severe forms (SMA Type 1), permanent ventilation or death in 90% of cases by age 2. It is caused by a lack of a functional survival motor neuron 1 (SMN1) gene, and in the most severe forms results in the rapid and irreversible loss of motor neurons, affecting muscle functions, including breathing, swallowing and basic movement.
Dr. Tauscher-Wisniewski,[/caption]
Sitra Tauscher-Wisniewski, MD
Vice President Clinical Development & Analytics
Novartis Gene Therapies
MedicalResearch.com: What is the background for this study? Would you briefly describe the condition of Spinal muscular atrophy (SMA)?
Response: At the 2023 Muscular Dystrophy Association Conference, we presented new data from two of our Long-Term Follow-Up (LTFU) studies, LT001 and LT002, which show the continued efficacy and durability of Zolgensma across a range of patient populations, with an overall benefit-risk profile that remains favorable. LT001 is a 15-year ongoing observational LTFU study following the Phase 1 START patients, who were the very first patients to receive our gene replacement therapy. LT-002 is a voluntary Phase 4 15-year ongoing follow-up safety and efficacy study of Zolgensma IV and investigational intrathecal (IT) OAV101 in patients previously treated in the Phase 3 IV studies (STR1VE-US, STR1VE-EU, STR1VE-AP, SPR1NT) and the Phase 1 IT study (STRONG).
Spinal muscular atrophy (SMA) is a rare, devastating genetic disease that leads to progressive muscle weakness, paralysis, and when left untreated in one of its most severe forms (SMA Type 1), permanent ventilation or death in 90% of cases by age 2. It is caused by a lack of a functional survival motor neuron 1 (SMN1) gene, and in the most severe forms results in the rapid and irreversible loss of motor neurons, affecting muscle functions, including breathing, swallowing and basic movement.
 Dr. Mapara[/caption]
Markus Y Mapara, MD
Professor of Medicine
Director of the Blood and Marrow Transplantation
Columbia University Medical Center
MedicalResearch.com: What is the background for this study? What are the main findings?
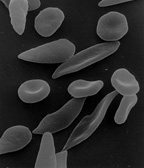
Response: Sickle cell disease is caused by a point mutation in the beta-globin gene of hemoglobin resulting in the production of abnormal hemoglobin which leads to formation of sickle-shaped RBC under conditions of low oxygen. Sickle cell disease affects about 100,000 patients in the US which are predominantly African American. The only curative approach is to perform an allogeneic bone marrow transplant which is however fraught with significant treatment-related risks if a matched sibling donor is not available.
The current study describes the successful application of a novel gene therapy to treat patients with sickle cell disease. The strategy is based on a gene-addition approach to introduce the genetic information for a Hemoglobin F-like molecule termed HgAT87Q into hematopoietic stem cells. The expression of this novel hemoglobin prevents polymerization of HgbS and has now been demonstrated to prevent the occurrence of vaso-occlusive pain crises in sickle cell disease patients.
Dr. Mapara[/caption]
Markus Y Mapara, MD
Professor of Medicine
Director of the Blood and Marrow Transplantation
Columbia University Medical Center
MedicalResearch.com: What is the background for this study? What are the main findings?
Response: Sickle cell disease is caused by a point mutation in the beta-globin gene of hemoglobin resulting in the production of abnormal hemoglobin which leads to formation of sickle-shaped RBC under conditions of low oxygen. Sickle cell disease affects about 100,000 patients in the US which are predominantly African American. The only curative approach is to perform an allogeneic bone marrow transplant which is however fraught with significant treatment-related risks if a matched sibling donor is not available.
The current study describes the successful application of a novel gene therapy to treat patients with sickle cell disease. The strategy is based on a gene-addition approach to introduce the genetic information for a Hemoglobin F-like molecule termed HgAT87Q into hematopoietic stem cells. The expression of this novel hemoglobin prevents polymerization of HgbS and has now been demonstrated to prevent the occurrence of vaso-occlusive pain crises in sickle cell disease patients.