Author Interviews, Infections, JAMA / 21.10.2020
Surveillance for Human Prion Disease
MedicalResearch.com Interview with:
Liliana Sanchez-Gonzalez MD, MPH
Medical Epidemiologist
Dengue Branch – Division of Vector Borne Diseases
Centers for Disease Control and Prevention
San Juan, PR
MedicalResearch.com: What is the background for this study? Would you briefly explain the significance of prion disease?
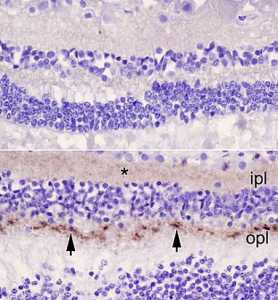
Response: Prion diseases are neurodegenerative diseases that occur in animals and humans. These diseases are caused by an infectious agent known as a prion. While the accuracy of diagnostic tests using cerebrospinal fluid or brain imaging from living patients has improved greatly in recent years, analysis of brain tissue is still necessary to confirm the diagnosis of these diseases.
Human prion disease cases are rare, but always fatal. There have been around 500 reported cases annually in the US in recent years. A very small percentage of human prion disease cases are acquired, meaning they are caused by an exposure to the infectious agent from an external source. The most well-known acquired human prion disease is variant Creutzfeldt-Jakob disease (vCJD), which was first described in the United Kingdom in 1996 and linked to consumption of contaminated beef from cattle with the animal prion disease bovine spongiform encephalopathy (BSE, or “mad cow” disease).
The only US state where classic BSE has been reported is Washington, where an infected dairy cow was imported from Canada in 2003. Beef from the slaughtered cow was processed for human consumption, and beef from cattle slaughtered the same day at the involved slaughter plant was recalled. After this incident, the Washington State Department of Health, in collaboration with the US Centers for Disease Control and Prevention and the National Prion Disease Pathology Surveillance Center (NPDPSC), implemented enhanced human prion disease surveillance. All patients with positive results from tests conducted at the NPDPSC are investigated. We present the results of 12 years of human prion disease surveillance, from 2006 to 2017, plus results of surveillance for vCJD through July 2020. (more…)