Posted at 20:59h
in
Author Interviews
MedicalResearch.com Interview with:
[caption id="attachment_47123" align="alignleft" width="184"]

Dr-Caughey[/caption]
Byron Caughey, Ph.D.
Senior Investigator
Chief, TSE/prion Biochemistry Section
Laboratory of Persistent Viral Diseases
NIH/NIAID Rocky Mountain Laboratories .
Hamilton, MT 59840 USA
MedicalResearch.com: What is the background for this study? Would you briefly explain the significance of prion-induced diseases and why they have been difficult to diagnosis?
Response: Although prion diseases such as Creutzfeldt-Jakob disease (CJD) are rarer than many other neurodegenerative diseases in humans, they are rapidly fatal, untreatable and transmissible. Therefore it is important to be able to diagnose prion diseases as early as possible, not only to accurately inform patients, families and caregivers, but also to reduce risks of transmission and improve prospects for developing therapeutics.
Toward these goals, we have shown that our
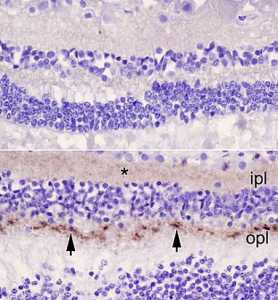
RT-QuIC prion seed amplification assays are highly accurate for diagnosing sporadic CJD using patients’ spinal fluid and/or nasal brushings in the clinical phase of disease. However, these particular specimens may not always be available, and it remains unclear how early they become RT-QuIC-positive in infected individuals in the months or years prior to the onset of overt clinical signs. We also showed recently that skin samples obtained
post-mortem from sCJD patients are RT-QuIC positive.
In the current study, we determined how early prion seeds appear in the rodents infected with prions in order to gain clues as to whether analyses of skin biopsies might provide a means of early preclinical detection of prion infections in humans.


 Dr. Haigh[/caption]
Cathryn Haigh, Ph.D.
Chief Prion Cell Biology Unit
Laboratory of Neurological Infections and Immunity
National Institute of Allergy and Infectious Diseases
Division of Intramural Research, Rocky Mountain Laboratories
National Institutes of Health
Hamilton, MT 59840
MedicalResearch.com: What is the background for this study, ie what are prions/prion-related diseases? Where are prions found?
Response: Prion diseases are infectious neurodegenerative diseases of humans and animals. In humans these diseases often manifest as rapidly progressing dementias but are rarely caused by a known exposure to the infectious agents (prions). More commonly they are sporadic (no known cause) or hereditary.
One form of human disease is believed to have arisen from eating beef contaminated with bovine spongiform encephalopathy (as known as mad cow disease). This has resulted in concerns that chronic wasting disease (CWD), a prion disease affecting deer, elk and moose, might also have the potential to cross the species barrier and cause disease in humans. To date, transmissions of CWD prions to cynomolgus macaques have been negative, a good sign that crossing the species barrier would not be easy, but macaques are not human so we wanted to test whether CWD could infect human brain tissue.
To do this we used a human cerebral organoid model (mini human brain tissues grown from skin cells in a laboratory) and directly exposed the organoids to prions from the brains of animals that had died of CWD.
Dr. Haigh[/caption]
Cathryn Haigh, Ph.D.
Chief Prion Cell Biology Unit
Laboratory of Neurological Infections and Immunity
National Institute of Allergy and Infectious Diseases
Division of Intramural Research, Rocky Mountain Laboratories
National Institutes of Health
Hamilton, MT 59840
MedicalResearch.com: What is the background for this study, ie what are prions/prion-related diseases? Where are prions found?
Response: Prion diseases are infectious neurodegenerative diseases of humans and animals. In humans these diseases often manifest as rapidly progressing dementias but are rarely caused by a known exposure to the infectious agents (prions). More commonly they are sporadic (no known cause) or hereditary.
One form of human disease is believed to have arisen from eating beef contaminated with bovine spongiform encephalopathy (as known as mad cow disease). This has resulted in concerns that chronic wasting disease (CWD), a prion disease affecting deer, elk and moose, might also have the potential to cross the species barrier and cause disease in humans. To date, transmissions of CWD prions to cynomolgus macaques have been negative, a good sign that crossing the species barrier would not be easy, but macaques are not human so we wanted to test whether CWD could infect human brain tissue.
To do this we used a human cerebral organoid model (mini human brain tissues grown from skin cells in a laboratory) and directly exposed the organoids to prions from the brains of animals that had died of CWD.