MedicalResearch.com Interview with:
[caption id="attachment_60529" align="alignleft" width="200"]

Dr. Hafezi-Moghadam[/caption]
Ali Hafezi-Moghadam, Ph.D., M.D
Director, Molecular Biomarkers Nano-Imaging Laboratory (MBNI)
Associate Professor of Radiology, Harvard Medical School
Brigham and Women’s Hospital
MedicalResearch.com: What is the background for this study?
Response: “It is very easy to answer many fundamental biological questions” said Richard Feynman in his 1959 address, where he also offered his simple and ingenious solution: “you just look at the thing!”
[1].
As a biologist, I am familiar with the challenges surrounding looking at things in the context of life. There is no single device or technology that lets me simply see the answers to my questions. How does diabetes harm the tissues in the body? When exactly does the pathology start and which molecules and cells are involved? Trying to answer these questions, I have spent the past two decades innovating new ways of quantifying expression of molecules in the living organism
[2].
At the same time to study diabetes, we needed a realistic rodent model that mirrors the human disease. In collaboration with KC Hayes
[3], we first introduced the Nile grass rat (NGR, Arvicanthis niloticus), a gerbil that recapitulates the main features of the human type 2 diabetes
[4]. For visualization of early changes, the eye offers a unique site. Much of my lab’s work focused on the first effects of diabetes in the retina, the site of the neurons that perceive light in the back of the eye
[5], [6], [7]. In recent studies, we focused on how diabetes affects the lens in the eye of our animals
[8], [9].
Diabetes is a major risk factor for cataract formation, a condition during which the lens loses its original transparency to visible light. How diabetic cataracts are formed is not well understood. A popular and prevailing theory, termed “sugar cataracts”, has been around for over half a century. According to the sugar hypothesis of cataracts, the excess levels of the sugar molecule, glucose, in the lens are transformed through the polyol pathway into the sugar-alcohol sorbitol. The resulting osmotic dysbalance leads to swelling of the fiber cells and opacity of the lens. Even though the sugar hypothesis has never been proven, it was generally accepted and remained unchallenged for a very long time. That is where our latest experimental results became relevant.







 Another name for LASIK is Laser-Assisted In Situ Keratomileusis, a popular vision correction surgery that has helped millions achieve more precise vision. It involves using advanced laser technology to modify the cornea to let light reach the eye and be correctly focused onto the retina. This reshaping process significantly improves vision for those with refractive defects, including astigmatism, farsightedness, and nearsightedness. If you've been considering options like
Another name for LASIK is Laser-Assisted In Situ Keratomileusis, a popular vision correction surgery that has helped millions achieve more precise vision. It involves using advanced laser technology to modify the cornea to let light reach the eye and be correctly focused onto the retina. This reshaping process significantly improves vision for those with refractive defects, including astigmatism, farsightedness, and nearsightedness. If you've been considering options like
 LASIK is an excellent option for people who have to wear glasses or contacts and don't want to do that anymore. Before going in for a consultation, it's good to learn as much as possible about it. Here are some answers, including ones if you have
LASIK is an excellent option for people who have to wear glasses or contacts and don't want to do that anymore. Before going in for a consultation, it's good to learn as much as possible about it. Here are some answers, including ones if you have 




 Dr. Singh[/caption]
Dr. Saundra Singh M.D., Ph.D.
Founder & CEO/President
Dr. Singh[/caption]
Dr. Saundra Singh M.D., Ph.D.
Founder & CEO/President
 MedicalResearch.com: What is the mission of
MedicalResearch.com: What is the mission of 
 Prof. Rahi[/caption]
Prof. Jugnoo S Rahi
Professor of Ophthalmic Epidemiology and Honorary Consultant Ophthalmologist
NIHR Senior Investigator
Head,
Prof. Rahi[/caption]
Prof. Jugnoo S Rahi
Professor of Ophthalmic Epidemiology and Honorary Consultant Ophthalmologist
NIHR Senior Investigator
Head, 
 Dr. Lee[/caption]
Cecilia S. Lee, MD, MS
Associate Professor,Director, Clinical Research
Department of Ophthalmology
Harborview Medical Center
University of Washington Seattle, WA
MedicalResearch.com: What is the background for this study?
Response: Cataract is a natural aging process of the eye and affects the majority of older adults who are at risk for dementia. Sensory loss, including vision and hearing, is of interest to the research community as a possible risk factor for dementia, and also as a potential point of intervention. Because cataract surgery improves visual function, we hypothesized that older people who undergo cataract surgery may have a decreased risk of developing Alzheimer disease and dementia.
We used the longitudinal data from an ongoing, prospective, community based cohort, Adult Changes in Thought (ACT) study. The ACT study includes over 5000 participants to date who are dementia free at recruitment and followed until they develop Alzheimer disease or dementia. We had access to their extensive medical history including comprehensive ophthalmology visit data. We investigated whether cataract surgery was associated with a decreased risk of developing Alzheimer disease and dementia.
Dr. Lee[/caption]
Cecilia S. Lee, MD, MS
Associate Professor,Director, Clinical Research
Department of Ophthalmology
Harborview Medical Center
University of Washington Seattle, WA
MedicalResearch.com: What is the background for this study?
Response: Cataract is a natural aging process of the eye and affects the majority of older adults who are at risk for dementia. Sensory loss, including vision and hearing, is of interest to the research community as a possible risk factor for dementia, and also as a potential point of intervention. Because cataract surgery improves visual function, we hypothesized that older people who undergo cataract surgery may have a decreased risk of developing Alzheimer disease and dementia.
We used the longitudinal data from an ongoing, prospective, community based cohort, Adult Changes in Thought (ACT) study. The ACT study includes over 5000 participants to date who are dementia free at recruitment and followed until they develop Alzheimer disease or dementia. We had access to their extensive medical history including comprehensive ophthalmology visit data. We investigated whether cataract surgery was associated with a decreased risk of developing Alzheimer disease and dementia. 
 Dr. Dalton[/caption]
Kristine Dalton PhD FAAO, FBCLA
School of Optometry & Vision Science
University of Waterloo
Waterloo, Canada
MedicalResearch.com: What is the background for this study?
Response: Dynamic visual acuity refers to the ability to detect and perceive small details in objects that are moving relative to an observer. Dynamic visual acuity is a complex visual function, that involves a number of different aspects of vision, including detecting the target, moving the eyes appropriately to observe the target, and processing the visual information from the target in the brain to interpret what we are seeing. What makes dynamic visual acuity so interesting to study, is that as a visual function, it appears to play an important role in a number of real-world situations, including playing sports, driving, and piloting, and it may provide us more information about how the visual system is functioning compared to the more traditional, static vision tests alone.
Previous research has demonstrated that consumption of caffeine has been shown to benefit physiological, psychomotor, and cognitive performance. More recently there has been an increased interest in studying the impacts of caffeine on the vision system, however the impact of caffeine on dynamic visual acuity has not been studied. This study was designed to address this limitation in the literature, particularly because dynamic visual acuity appears to be such an important visual function for real-world activities.
Dr. Dalton[/caption]
Kristine Dalton PhD FAAO, FBCLA
School of Optometry & Vision Science
University of Waterloo
Waterloo, Canada
MedicalResearch.com: What is the background for this study?
Response: Dynamic visual acuity refers to the ability to detect and perceive small details in objects that are moving relative to an observer. Dynamic visual acuity is a complex visual function, that involves a number of different aspects of vision, including detecting the target, moving the eyes appropriately to observe the target, and processing the visual information from the target in the brain to interpret what we are seeing. What makes dynamic visual acuity so interesting to study, is that as a visual function, it appears to play an important role in a number of real-world situations, including playing sports, driving, and piloting, and it may provide us more information about how the visual system is functioning compared to the more traditional, static vision tests alone.
Previous research has demonstrated that consumption of caffeine has been shown to benefit physiological, psychomotor, and cognitive performance. More recently there has been an increased interest in studying the impacts of caffeine on the vision system, however the impact of caffeine on dynamic visual acuity has not been studied. This study was designed to address this limitation in the literature, particularly because dynamic visual acuity appears to be such an important visual function for real-world activities. 





 Dr. Walline[/caption]
Jeffrey J. Walline, OD PhD
Associate Dean for Research
The Ohio State University
Columbus, OH 43210-1240
MedicalResearch.com: What is the background for this study?
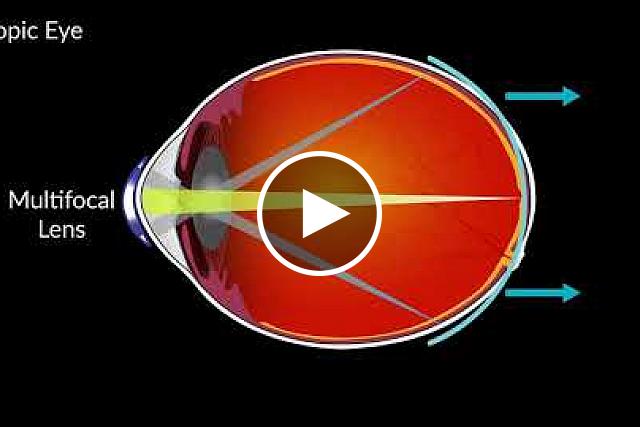
Response: Greater amounts of nearsightedness are related to higher risks of sight-threatening complications in adulthood, so anything we can do to slow the progression of nearsightedness in childhood can have meaningful benefits in the future.
As the prevalence of nearsightedness increases worldwide and affects approximately 1/3 of the people in the United States, a treatment that provides clear vision AND slows the progression of nearsightedness can have a profound effect.
Dr. Walline[/caption]
Jeffrey J. Walline, OD PhD
Associate Dean for Research
The Ohio State University
Columbus, OH 43210-1240
MedicalResearch.com: What is the background for this study?
Response: Greater amounts of nearsightedness are related to higher risks of sight-threatening complications in adulthood, so anything we can do to slow the progression of nearsightedness in childhood can have meaningful benefits in the future.
As the prevalence of nearsightedness increases worldwide and affects approximately 1/3 of the people in the United States, a treatment that provides clear vision AND slows the progression of nearsightedness can have a profound effect.



 Prof. FAN Zhiyong PhD
University of California, Irvine
HKUST School of Engineering
MedicalResearch.com: What is the background for this study? What are the main findings?
Response: According to the report of The World Health Organization, there are over 252 million people suffering from visual impairment globally and 15 million of them are difficult to cure by conventional medical methods. However, today, even the best bionic eyes have only 200 clinical trials, less than 1 ppm of all the patients, mainly due to their poor performance and high cost. The huge gap in supply and demand triggers the study of bionic eyes with performance comparable to human eyes. One important reason for their poor performance is the mismatch in shape between the flat bionic eyes and concave sclera. To protect the soft tissue in eyes from being damaged by the bionic surface, the implanted bionic eyes have to be small. This has limited the sensing area and further the electrodes number, and finally yielded poor image sensing characters with low resolution and narrow field-of-view.
In this work, we are trying to achieve high performance image sensing by biomimeticing human eyes. The high-density NWs are well aligned and embedded in a hemispherical template to serve as retina. The conformal attachment of bionic eyes with sclera enables the large sensing area and wide visual angle. In addition, each individual high-density nanowires can potentially work as an individual pixel. By addressing these challenges, our device design has huge potential to improve the image sensing performance of bionic eyes.
Prof. FAN Zhiyong PhD
University of California, Irvine
HKUST School of Engineering
MedicalResearch.com: What is the background for this study? What are the main findings?
Response: According to the report of The World Health Organization, there are over 252 million people suffering from visual impairment globally and 15 million of them are difficult to cure by conventional medical methods. However, today, even the best bionic eyes have only 200 clinical trials, less than 1 ppm of all the patients, mainly due to their poor performance and high cost. The huge gap in supply and demand triggers the study of bionic eyes with performance comparable to human eyes. One important reason for their poor performance is the mismatch in shape between the flat bionic eyes and concave sclera. To protect the soft tissue in eyes from being damaged by the bionic surface, the implanted bionic eyes have to be small. This has limited the sensing area and further the electrodes number, and finally yielded poor image sensing characters with low resolution and narrow field-of-view.
In this work, we are trying to achieve high performance image sensing by biomimeticing human eyes. The high-density NWs are well aligned and embedded in a hemispherical template to serve as retina. The conformal attachment of bionic eyes with sclera enables the large sensing area and wide visual angle. In addition, each individual high-density nanowires can potentially work as an individual pixel. By addressing these challenges, our device design has huge potential to improve the image sensing performance of bionic eyes.

