MedicalResearch.com Interview with:
[caption id="attachment_60763" align="alignleft" width="200"]

Dr. Worthley[/caption]
Daniel L. Worthley
MBBS (Hons), PhD, MPH, FRACP, AGAF
Gastroenterologist
Associate Professor
University of Adelaide
MedicalResearch.com: What is the background for this study?
Response: Cells are revolutionising healthcare, from modern faecal microbial transplantation in the gut to CAR-T cells fighting cancers, life healing life. Some aspects of cellular care are so entrenched in medicine that they are almost overlooked for the miraculous cellular therapies that they are, such as stem cell transplantation to treat haematological malignancies and, of course, in vitro fertilization, life creating life. Modern medicine is slowly, but surely, pivoting from pills to cells. Professor Siddhartha Mukherjeee, oncologist, scientist, and author, provides a beautiful thesis of this in his book Song of the Cell and in his TED talk on the cellular revolution in medicine (
https://youtu.be/qG_YmIPFO68?feature=shared). I was lucky enough to have trained with Sid as a post-doc at Columbia and this concept was really drummed into me. But, as a gastroenterologist, perhaps it was the bacterial cells, rather than the blood cells, that had most to offer in the management of bowel disorders? Around the same time, Professors Jeff Hasty, Tal Danino and Omar Din from UC San Diego had been inventing and publishing, in my opinion, the best bacterial engineering work that has ever been produced to specifically target cancer. I remember when we first reviewed their 2016 Nature paper in our lab meeting (
https://www.nature.com/articles/nature18930#citeas), it was like – “We gotta meet these guys!”. Through Tal, who was by then, working at Columbia, I was introduced to Jeff and I attended his lab meeting back in 2019. That was where our project began after a lab meeting in La Jolla. Rob Cooper had presented his work on horizontal gene transfer. Everything that comes out of Jeff’s lab is both practical and reproducible but also beautiful. Beautiful in a scientific self-evident way that instantly communicates the purpose, approach and outcomes of an experiment.
Rob’s presentation that day was a case-in-point. Rob was studying genes and gene transfer in bacteria (see part of Rob’s fascinating presentation here,
https://youtu.be/5nBsRF-BsA8?feature=shared). Genes are the fundamental unit of heredity and gene transfer (or inheritance) the process by which genes are passed from one cell to another. Genes may be inherited vertically when one cell replicates its DNA and divides into two, now separate, cells (reproduction). Genes are the stuff, and vertical gene transfer is the process, by which you receive your mother’s laugh and your father’s eyebrows. Genes may also, however, be inherited horizontally when DNA is passed between unrelated cells, outside of parent to offspring inheritance. Horizontal gene transfer is quite common in the microbial world. Certain bacteria can salvage genes from cell-free DNA found within its environment. This sweeping up of cell-free DNA, into a cell, is called natural competence. So, competent bacteria can sample their nearby environment and, in doing so, acquire genes that may provide a selective advantage to that cell. Like cellular panning for flecks of gold in a stream. After Rob’s presentation, Jeff, Rob and I started to discuss the possibilities. If bacteria can take up DNA, and cancer is defined genetically by a change in its DNA then, theoretically, bacteria could be engineered to detect cancer. Colorectal cancer seemed a logical proof of concept as the colorectal lumen is full of microbes and, in the setting of cancer, full of tumour DNA. When a biophysicist, a scientist and a gastroenterologist walk into a bar, after a lab meeting, this is what can happen! Professor Susi Woods and Dr Josephine Wright, superb cancer scientists from Adelaide, Australia, were quickly recruited in as essential founding members of the group. We all got to work. Australian and US grants, lots of experiments, early morning Zoom calls across the Pacific, inventing new animal models and approaches, i.e. a many year, iterative process of design-build-test-learn, that got us all to where we are now.







 Dr. Zeynep Gümüş[/caption]
Zeynep H. Gümüş, PhD
Associate Professor
Icahn School of Medicine at Mount Sinai
MedicalResearch.com: What is the background for this study?
Response: The germline genome of each individual person has a unique combination of millions of genetic variants that influence virtually all biological processes throughout life, including cancer evolution. In this study, we have investigated the impact of germline variants – genetic defects one is born with – on gene expression and protein abundance in tumors across cancer types.
MedicalResearch.com: Would you describe the technique of precision peptidomics?
Response: We have leveraged a cohort of 1,064 patients with multiple cancer types to explore the impact of germline variations on cancer-relevant genes through multiple-omics layers: from DNA to RNA, protein abundance and post-translational modifications. To assess the effects of coding variants and their association with cognate proteins, we used precision peptidomics, which is the quantification of peptides carrying genetic variants from individual patients. Through this approach, we mapped 337,469 protein coding germline variants onto patient peptides, revealing their potential impact on protein modifications, protein stability, allele-specific expression, and protein structure by leveraging the relevant protein databases.
Dr. Zeynep Gümüş[/caption]
Zeynep H. Gümüş, PhD
Associate Professor
Icahn School of Medicine at Mount Sinai
MedicalResearch.com: What is the background for this study?
Response: The germline genome of each individual person has a unique combination of millions of genetic variants that influence virtually all biological processes throughout life, including cancer evolution. In this study, we have investigated the impact of germline variants – genetic defects one is born with – on gene expression and protein abundance in tumors across cancer types.
MedicalResearch.com: Would you describe the technique of precision peptidomics?
Response: We have leveraged a cohort of 1,064 patients with multiple cancer types to explore the impact of germline variations on cancer-relevant genes through multiple-omics layers: from DNA to RNA, protein abundance and post-translational modifications. To assess the effects of coding variants and their association with cognate proteins, we used precision peptidomics, which is the quantification of peptides carrying genetic variants from individual patients. Through this approach, we mapped 337,469 protein coding germline variants onto patient peptides, revealing their potential impact on protein modifications, protein stability, allele-specific expression, and protein structure by leveraging the relevant protein databases.


 Dr. Nas[/caption]
Dr Zeynep Nas Ph.D.
Postdoctoral Research Fellow
Department of Behavioural Science and Health
Institute of Epidemiology & Health Care
University College London
MedicalResearch.com: What is the background for this study?
Response: We were interested in why some children are more selective in their food intake and more reluctant to try new foods compared to those who are not. We investigated this question in a twin study, which compares identical twins (who share all of their genes) to non-identical twins (who share half) to understand the relative influence of genetics versus the environment in shaping individual differences in fussy eating.
Dr. Nas[/caption]
Dr Zeynep Nas Ph.D.
Postdoctoral Research Fellow
Department of Behavioural Science and Health
Institute of Epidemiology & Health Care
University College London
MedicalResearch.com: What is the background for this study?
Response: We were interested in why some children are more selective in their food intake and more reluctant to try new foods compared to those who are not. We investigated this question in a twin study, which compares identical twins (who share all of their genes) to non-identical twins (who share half) to understand the relative influence of genetics versus the environment in shaping individual differences in fussy eating.

 Dr. Walsh[/caption]
Christopher Walsh, M.D., Ph.D.
Chief, Division of Genetics and Genomics
Bullard Professor of Pediatrics and Neurology at Harvard Medical School
and researcher who has used material donated to the brain bank
MedicalResearch.com: What is the background for this study?
Response: Many different types of genetic variants contribute to neurodevelopmental disorders such as autism. Copy number variants are large pieces of genetic material that are duplicated or deleted. We have known for many years that many copy number variants at certain genetic locations are linked to autism. Because these copy number variants may include lots of different genes, it has been difficult to understand how these copy number variants alter human brain function. Furthermore, although animal models are important, autism is in many ways defined by differences in uniquely human cognitive and social functioning. Better understanding of how these copy number variants change human brain function will shed light on universal mechanisms that regulate neurodevelopment. We studied a copy number variant called dup15q, that is associated with almost 40-fold higher rates of autism vs. the general population. We studied post-mortem human brain tissue from individuals with dup15q, individuals with autism not related to dup15q, and neurotypical controls, to better understand how the human brain is impacted by dup15q. We focused on frontal cortex, an important brain region in executive function and social perspective taking. We applied cutting edge techniques that allow us to assess individual cells in the brain.
Dr. Walsh[/caption]
Christopher Walsh, M.D., Ph.D.
Chief, Division of Genetics and Genomics
Bullard Professor of Pediatrics and Neurology at Harvard Medical School
and researcher who has used material donated to the brain bank
MedicalResearch.com: What is the background for this study?
Response: Many different types of genetic variants contribute to neurodevelopmental disorders such as autism. Copy number variants are large pieces of genetic material that are duplicated or deleted. We have known for many years that many copy number variants at certain genetic locations are linked to autism. Because these copy number variants may include lots of different genes, it has been difficult to understand how these copy number variants alter human brain function. Furthermore, although animal models are important, autism is in many ways defined by differences in uniquely human cognitive and social functioning. Better understanding of how these copy number variants change human brain function will shed light on universal mechanisms that regulate neurodevelopment. We studied a copy number variant called dup15q, that is associated with almost 40-fold higher rates of autism vs. the general population. We studied post-mortem human brain tissue from individuals with dup15q, individuals with autism not related to dup15q, and neurotypical controls, to better understand how the human brain is impacted by dup15q. We focused on frontal cortex, an important brain region in executive function and social perspective taking. We applied cutting edge techniques that allow us to assess individual cells in the brain. 


 Ben Petrazzini[/caption]
Ben Omega Petrazzini, B.Sc.
Associate Bioinformatician
Ben Petrazzini[/caption]
Ben Omega Petrazzini, B.Sc.
Associate Bioinformatician



 Dr. Belloy[/caption]
Michael E. Belloy, PhD
Department of Neurology and Neurological Sciences
Stanford University, Stanford, California
MedicalResearch.com: What is the background for this study?
Response: Apolipoprotein E (APOE)*2 and APOE*4 are, respectively, the strongest protective and risk-increasing, genetic variants for late-onset Alzheimer disease. As such, one’s APOE genotype is highly relevant towards clinical trial design and Alzheimer’s disease research. However, most insights so far are focused on the associations of these APOE genotypes with Alzheimer’s disease risk in non-Hispanic white individuals.
One important aspect of our work is that we really increased sample sizes for non-Hispanic Black, Hispanic, and East Asian individuals, so that we now have better understanding of the associations of APOE genotypes with Alzheimer’s disease risk in these groups. In complement, we also did the largest investigation to date on the role of ancestry on the associations of APOE genotypes with Alzheimer’s disease risk. The scale of our study was thus a critical factor in generating novel insights.
Dr. Belloy[/caption]
Michael E. Belloy, PhD
Department of Neurology and Neurological Sciences
Stanford University, Stanford, California
MedicalResearch.com: What is the background for this study?
Response: Apolipoprotein E (APOE)*2 and APOE*4 are, respectively, the strongest protective and risk-increasing, genetic variants for late-onset Alzheimer disease. As such, one’s APOE genotype is highly relevant towards clinical trial design and Alzheimer’s disease research. However, most insights so far are focused on the associations of these APOE genotypes with Alzheimer’s disease risk in non-Hispanic white individuals.
One important aspect of our work is that we really increased sample sizes for non-Hispanic Black, Hispanic, and East Asian individuals, so that we now have better understanding of the associations of APOE genotypes with Alzheimer’s disease risk in these groups. In complement, we also did the largest investigation to date on the role of ancestry on the associations of APOE genotypes with Alzheimer’s disease risk. The scale of our study was thus a critical factor in generating novel insights.
 Dr. Stark[/caption]
Dr Mitchell Stark
Dr. Stark[/caption]
Dr Mitchell Stark
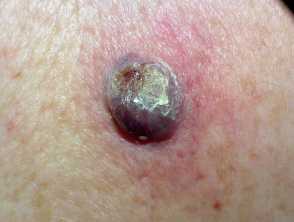
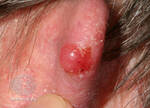 One example of a nodular melanoma without pigment.
One example of a nodular melanoma without pigment. 


 Response: We were broadly interested in discovering instances of bacterial genes that have been acquired by diverse animal genomes over millions of years of evolution by the process of horizontal gene transfer (HGT). Since these events are quite rare and most previous discoveries have been serendipitous, we developed computational methods to identify genes acquired by HGT in animals. One of the exciting discoveries from our work was that vertebrate IRBP appeared to have originated in bacteria and is now a critical component of the vertebrate visual cycle, so this paper focuses on that one discovery.
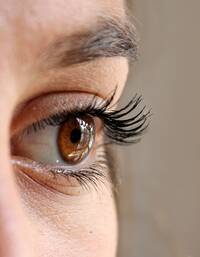
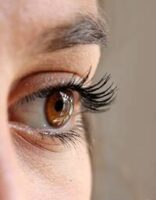
IRBP or interphotoreceptor retinoid binding protein is an important protein present in the space between two major cell types in our eyes, photoreceptor cells and RPE cells. Our ability to see involves an intricate set of steps where light is first sensed by causing a change (isomerization) in the chemical structure of molecules in the eye called retinoids. This sensing of light occurs in our photoreceptor cells. Following this change in the chemical structure, the retinoid needs to be recycled back to the chemical structure that can again sense light. This recycling occurs in RPE cells. IRBP performs the essential function of shuttling retinoids between the photoreceptors and the RPE cells, which allows the cycle of sensing and regeneration to work. Supporting its importance, mutations in IRBP (also known as retinol binding protein 3 or RBP3) can cause several severe human eye diseases.
Response: We were broadly interested in discovering instances of bacterial genes that have been acquired by diverse animal genomes over millions of years of evolution by the process of horizontal gene transfer (HGT). Since these events are quite rare and most previous discoveries have been serendipitous, we developed computational methods to identify genes acquired by HGT in animals. One of the exciting discoveries from our work was that vertebrate IRBP appeared to have originated in bacteria and is now a critical component of the vertebrate visual cycle, so this paper focuses on that one discovery.
IRBP or interphotoreceptor retinoid binding protein is an important protein present in the space between two major cell types in our eyes, photoreceptor cells and RPE cells. Our ability to see involves an intricate set of steps where light is first sensed by causing a change (isomerization) in the chemical structure of molecules in the eye called retinoids. This sensing of light occurs in our photoreceptor cells. Following this change in the chemical structure, the retinoid needs to be recycled back to the chemical structure that can again sense light. This recycling occurs in RPE cells. IRBP performs the essential function of shuttling retinoids between the photoreceptors and the RPE cells, which allows the cycle of sensing and regeneration to work. Supporting its importance, mutations in IRBP (also known as retinol binding protein 3 or RBP3) can cause several severe human eye diseases.



 Dr. Kleiman[/caption]
Norman Kleiman, PhD, MS
Department of Environmental Health Sciences
Mailman School of Public Health
Columbia University, New York, NY
MedicalResearch.com: What is the background for this study?
Response: The 1986 Chornobyl nuclear disaster caused the evacuation of 300,000 persons from the cities and villages surrounding the nuclear power plant complex. Pets and belongings were left behind, and the Soviet authorities ordered all animals within the Chornobyl Exclusion Zone killed. Some dogs evaded destruction, and some 300+ descendants of these animals live primarily at two locations today, immediately surrounding the Nuclear Power Plant (NPP) complex and about 10 km away in Chornobyl city. What is relatively unknown to the general public is that Chornobyl is not a desolate, abandoned wasteland. Some thousands of individuals work there every day in continuing cleanup activities and at two new fuel reprocessing facilities built near the damaged reactor. These areas have been substantially remediated, and the average radiation levels are relatively modest. The dogs, which, while feral, are accustomed to human interaction, live near the workers and are not currently exposed to high radiation levels. In contrast to lower radiation levels, there is a toxic mixture of heavy metals, organics, pesticides, and unknown chemicals left over from years’ long cleanup efforts and the decay of a large former military-industrial complex at the NPP.
Since 2016, the NPP authorities have brought in teams of veterinarians and volunteers to spay, neuter, and vaccinate the dogs to protect the workers and deal with a growing population. At the same time, some scientists joined the teams to obtain various kinds of biospecimens (hair, urine, feces, blood, saliva, parasites) to examine the animals’ health and learn how this toxic environment may have affected them or their offspring. Since dogs are human companion animals and live closely with us, any information we learn about health risks to the dogs may be relevant to protecting human workers and inform us about the kinds of health risks posed by ecological and environmental disasters in the future.
Dr. Kleiman[/caption]
Norman Kleiman, PhD, MS
Department of Environmental Health Sciences
Mailman School of Public Health
Columbia University, New York, NY
MedicalResearch.com: What is the background for this study?
Response: The 1986 Chornobyl nuclear disaster caused the evacuation of 300,000 persons from the cities and villages surrounding the nuclear power plant complex. Pets and belongings were left behind, and the Soviet authorities ordered all animals within the Chornobyl Exclusion Zone killed. Some dogs evaded destruction, and some 300+ descendants of these animals live primarily at two locations today, immediately surrounding the Nuclear Power Plant (NPP) complex and about 10 km away in Chornobyl city. What is relatively unknown to the general public is that Chornobyl is not a desolate, abandoned wasteland. Some thousands of individuals work there every day in continuing cleanup activities and at two new fuel reprocessing facilities built near the damaged reactor. These areas have been substantially remediated, and the average radiation levels are relatively modest. The dogs, which, while feral, are accustomed to human interaction, live near the workers and are not currently exposed to high radiation levels. In contrast to lower radiation levels, there is a toxic mixture of heavy metals, organics, pesticides, and unknown chemicals left over from years’ long cleanup efforts and the decay of a large former military-industrial complex at the NPP.
Since 2016, the NPP authorities have brought in teams of veterinarians and volunteers to spay, neuter, and vaccinate the dogs to protect the workers and deal with a growing population. At the same time, some scientists joined the teams to obtain various kinds of biospecimens (hair, urine, feces, blood, saliva, parasites) to examine the animals’ health and learn how this toxic environment may have affected them or their offspring. Since dogs are human companion animals and live closely with us, any information we learn about health risks to the dogs may be relevant to protecting human workers and inform us about the kinds of health risks posed by ecological and environmental disasters in the future.





